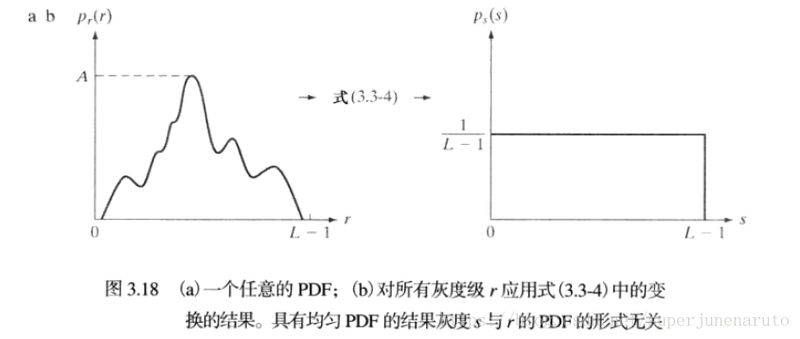
引言
在上一章中《第四章:Hatree-Fock方程 | 2026新版VASP基础教程》,我们系统学习了Hartree-Fock方法,理解了它如何通过单电子近似构建波函数并数值求解,为量子化学计算奠定了重要基石。然而,Hartree-Fock方法在处理电子相关效应时存在固有局限,且计算量随体系增大而急剧增长,难以高效描述复杂材料体系。
本章将正式引入现代第一性原理计算的核心——密度泛函理论。我们将从基本思想出发,阐述如何将多电子体系的能量表示为电子密度的泛函,并重点讲解Kohn-Sham方程的构建、交换关联泛函的物理意义与常见形式。
密度泛函理论
密度泛函理论是一种研究多电子体系(如原子、分子和固体)电子结构的量子力学方法。它是目前化学、物理和材料科学中最常用的从头算(ab initio)计算方法之一,已成为这些领域不可或缺的工具。
核心思想:从波函数到电子密度
传统的电子结构计算方法(如Hartree-Fock方法)通常依赖于波函数来描述系统状态。对于包含 N 个电子的体系,波函数是 3N 维的函数,计算难度极大。DFT 的核心创新在于引入了电子密度(density)这一概念。
定义
电子密度是一个只依赖于三个空间坐标(x,y,z)的函数,描述了空间中电子的分布情况。
优势
相比于波函数,电子密度只需要处理三个变量,显著降低了计算的复杂度。
基础定理
DFT 基于霍亨伯格-孔恩(Hohenberg-Kohn)定理,该定理表明电子密度与外部势能之间存在唯一的一一对应关系。这意味着,若已知基态的电子密度,就能确定体系的所有物理性质,反之亦然。
计算框架:Kohn-Sham 方法
尽管密度泛函理论简化了问题,但直接从电子密度计算能量仍然困难。为了解决这个问题,Kohn和Sham提出了Kohn-Sham方程,成为现代DFT计算的标准框架。
思想
Kohn-Sham 方法假设存在一个虚构的“非相互作用电子系统”,该系统具有与真实系统相同的基态电子密度。
核心方程
通过求解一组类似于Hartree-Fock的单电子方程(Kohn-Sham方程),可以得到电子密度和体系能量。
能量表达
体系的总能量被分解为若干部分,其中最关键的是交换-关联能(Exchange-Correlation Energy,XC能)。XC能包含了所有复杂的电子-电子相互作用效应,是理论的核心难点。
交换-关联泛函(XC Functional)
由于精确的XC能未知,实际计算中必须使用近似模型,这些模型统称为泛函(Functional)。
LDA(局部密度近似)
假设体系在每一点的电子密度与均匀电子气体相同。LDA是最早的近似,计算效率高,但精度有限。
GGA(广义梯度近似)
在LDA基础上,引入了电子密度梯度的修正。GGA显著改善了分子几何结构和反应能的预测精度,是目前最常用的近似之一。
Meta-GGA和混合泛函(Hybrid Functionals)
进一步引入了动能密度或将Hartree-FFock的精确交换项混入GGA中,以提高精度。例如著名的B3LYP泛函。
挑战
尽管近似不断改进,但新泛函往往在提高能量计算精度时,可能会牺牲电子密度的准确性,这在激发态计算中尤为突出。
主要应用领域
密度泛函理论因其计算效率高(相较于波函数方法)和精度较好,已广泛应用于多个科研和工业领域。
材料科学
用于预测材料的电子结构、带隙、磁性和光学性质。是设计新型半导体、光电材料的首选工具。
计算化学
研究分子的几何结构、振动频率、反应机理、吸附能和光谱性质。广泛用于药物设计和有机合成的机理分析。
催化剂研究
尤其在电催化和光催化领域,DFT用于揭示反应机理、计算反应能垒、筛选高活性催化剂(如水煤气变换、CO2还原等)。
电化学与电催化
用于模拟半导体催化剂和电化学界面,解决能带弯曲和电子转移问题,结合机器学习加速新材料发现。
气体传感器
模拟气体分子与传感材料表面的相互作用,设计高灵敏度的气体传感器。
现代进展与挑战
尽管DFT已经非常成熟,但在处理特定复杂体系时仍面临挑战,科研人员正积极探索新的解决方案。
多态密度泛函理论(MSDFT)
针对传统DFT在处理激发态、强关联系统(如过渡金属化合物)时的失效,MSDFT引入了矩阵密度作为基本变量。它通过非正交态相互作用(NOSI)算法和最小活性空间(MAS)概念,成功解决了激发态计算中的根本性困难。
机器学习整合
结合DFT计算生成的大规模数据,利用机器学习(ML)建立结构-性能关系模型。通过预测d带中心、吸附能等关键参数,大幅加速了催化剂筛选和新材料的发现。
高精度计算
随着计算能力提升,研究者致力于开发更高精度的泛函和方法,以解决如vdW相互作用(范德华力)和电子-电洞耦合等微观效应。
本章要点总结
密度泛函理论知识点梳理
基本思想:介绍了电子密度、霍亨伯格-孔恩定理
Khon-Sham方程:介绍了思想、核心方程、能量表达
交换关联泛函:介绍了LDA、GGA、Meta-GGA关联能
下一步学习建议
下一章将正式引入本次教程的核心—密度泛函理论。我们将从基本思想、Khon-Sham方程、交换关联泛函等角度出发,详细介绍密度发函理论,以及它在处理不同材料体系时的独特优势的应用,敬请期待!